実験の各段階で、材料の準備から始まり、PCRを継続し、シーケンスを終了すると、エラーの蓄積が発生します。 結果の重要性を評価するメカニズムが必要です。 ゲノムの特定の部分で見つかったリードが偶然ではない可能性はどのくらいですか? この記事で紹介するアプローチは、DNA-seqを使用して取得したデータに適用可能であり、ポアソン分布を使用して有意性を評価する可能性について説明しています。
この図は、配列決定のための材料の準備プロセスを概略的に示しています。 各段階で間違いが発生する可能性があり、その結果、ゲノムの任意の部分が読み取られます。
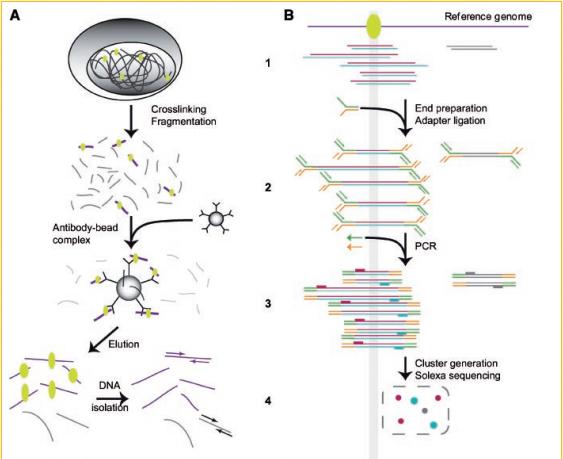
- A.1 DNA細胞は、タンパク質をよりよく固定するためにホルムアルデヒドで処理されます。 次に、 超音波を含むさまざまな方法でDNAを断片に切断します。
- A.2-3タンパク質に敏感な抗体が追加されます。 タンパク質でマークされたフラグメントが抗体に付着します。 一部のフラグメントはランダムに結合します。
- A.4 DNA断片による抗体の沈殿が起こります。 抗体およびタンパク質からのDNAフラグメントの精製。
- B.1-4これらのパラグラフは、得られたDNA断片を配列決定のために準備する手順を示しています。 特別なアダプターの追加。 PCRを使用した乗算。 スライドに配置します。
結果のスライドは、
Illuminaなどのシーケンスマシンに配置されます。 機器とそれに付属するプログラムの操作の結果は、FASTA / FASTQファイルです。 FASTQファイルの内容の例を次に示します。
@HWI-XXXXX_0470:1:1:1761:946
「@」で始まる1行目と「+」で始まる3行目は説明的なもので、名前、スライド番号などが含まれています。 2行目はヌクレオチドの読み取りシーケンスで、この例では36個あり、4行目はヌクレオチドを読み取る確率です。 以下は、ファイル形式の正確な説明と
http://en.wikipedia.org/wiki/FASTQ_formatのアルファベット表記から数値を取得する方法へのリンクです。
1回のシーケンスで何百万ものすべての読み取りを含むファイルは、現在最速の
ボウタイプログラムに転送されます。 ボウタイプログラムの実行行は次のとおりです。
/usr/src/bowtie-0.12.7/bowtie -q -v 2 -m 1 --best --strata -p 8 -S \ /usr/src/bowtie-0.12.7/indexes/hg19 - | samtools view -Sb - >${DIRNAME}/${SEQNAME}.bam
例で指定されたパラメーターは、次のプログラムの動作を特徴付けます。
-q-入力ファイルをFASTAではなくFASTQとして扱います
-v-読み取りで許可されるエラーの数。上記の例では2
-m-検索するゲノム内のエントリ(読み取り)の数、この場合1
-p-使用するスレッドの数。プロセッサコアの数から進みます-8
-S-出力ファイルが.sam形式で書き込まれることを意味します
/usr/src/bowtie-0.12.7/indexes/hg19-事前にインデックス付けされたゲノムへのパス。bowtie-buildによって実行されます。 また、完成したパスはインターネットからダウンロードできます。
-(2行目の最後のダッシュ)は、samtoolsプログラムに送信するための標準出力への出力を意味し、その場でそれを.bamに変換します。
説明では、ボウタイ操作の特別なモードを設定するパラメーター--best --strataをスキップしました:プログラムが異なるエラー数でゲノム内の読み取りの複数のオカレンスを検出した場合、エラーが最小の結果を表示します。 エラーが最小の結果が複数ある場合、それらは表示されません。 これにより、プログラムの動作時間が長くなりますが、同時にゲノムの読み取りの感度を上げることができます。 .sam / .bamファイルからの出力例を次に示します。
@HD VN:1.0 SO:unsorted @SQ SN:chr10 LN:135374737 @SQ SN:chr11 LN:134452384 … @PG ID:Bowtie VN:0.12.7 CL:"/usr/src/bowtie-0.12.7/bowtie -q -v 1 -m 1 --best --strata -p 8 -S" USI-EAS21_8_3445_7_1_452_540 0 chr5 39195556 255 23M * 0 0 CTCCAGATTCTCTGGAAAGATGG SSSSSSPSSSSSSSSGNNSJSSL XA:i:0 MD:Z:23 NM:i:0 USI-EAS21_8_3445_7_1_170_242 0 chr14 73509925 255 23M * 0 0 CTGCATTAGACCTAGGCTTAGAA SSSSSSSSSSSSSSSSSSSGSNN XA:i:0 MD:Z:23 NM:i:0 USI-EAS21_8_3445_7_1_290_985 0 chr7 142613233 255 23M * 0 0 AGCTGACTGGCAAGCAACAGAGT JSSSSSSSSSSSPSSGSSLSNSL XA:i:0 MD:Z:23 NM:i:0 USI-EAS21_8_3445_7_1_881_711 4 * 0 0 * * 0 0 ATCGGAAGAGCTCGTATGCCGTC SSSSPSSSSSSSSSSSSSSSSNS XM:i:0 USI-EAS21_8_3445_7_1_897_318 0 chr3 50101427 255 23M * 0 0 GGGCGCAGAACCGCTGCTGCTGC SPSSSSSSSPPSSSSNSPPPLSL XA:i:1 MD:Z:15A7NM:i:1
ヘッダー(@ SQ、@ HD、@ PGで始まる行)には説明が含まれています。染色体の名前とその長さ、ファイルがソートされているかどうか、ファイルを受信したコマンド。 ボディは行で構成され、各行は読み取りですが、ゲノムの座標を持ちます。 詳細については、
http://genome.sph.umich.edu/wiki/SAMをご覧
ください 。
ゲノム上の分布は異なる可能性がありますが、富裕地域では、分布が次の特性を備えている場合、実験が成功する可能性が高いと考えられています。 リードがゲノムをどのようにカバーできるかの例は、上の図に描かれています。
充実したサイトを決定するために、次の推論を使用します。 染色体のすべての長さを追加し、繰り返しセクションの長さ(hg19の場合は約2282603114)を減算し、結果の長さをGにします。長さGを幅がWのウィンドウ(たとえば500)に分割します。 Nに対して結果ウィンドウの数を取ります(G / W約4565206.228)。 ゲノム内で正常に検出された実験の読み取り数を計算し(合計4.5E6のうち4E6とします)、その数をSとします。その後、ウィンドウごとの平均読み取り数
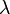
なります
= S * W / G = S / N
ポアソン分布

イベントの既知の平均発生源を使用して、一定期間におけるイベントの所定数kの発生源の確率を表します。 累積ポアソン分布は、kまでのイベントの発生確率を表します。 累積分布を使用します。これは、ウィンドウ内の読み取り回数がkより少ない確率に関心があるためです。 帰無仮説:kはウィンドウをランダムに読み取ります。 対立仮説:ウィンドウ内のk個のリードは偶発的ではありません。 帰無仮説が真である確率は偶数です
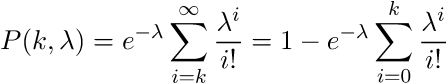
。 近いP(k、
)を0にすると、読み取りが所定のウィンドウに誤ってヒットする可能性が低くなります。 この例では、ウィンドウの読み取り回数が最大15回の場合、1-P(15.0.876193)= 2.55351E-15、つまり 幅が500のウィンドウでは、2.55351E-15の確率で最大15の読み取り数が偶然取得できます。 この数値はp値とも呼ばれます。 したがって、15を超える読み取りがある場合、それらが誤ってウィンドウに陥る可能性はさらに低くなります。 したがって、プログラムで条件を設定できます。p値<= 1.e16の場合、ウィンドウに入ったデータは考慮のために残され、残りは破棄されます。
以下は、
前の記事で説明したクラスの一部を使用し、新しいPoissonHandlerクラスによって補足されるサンプルプログラムです。
最初のアーカイブの追加ファイルは
ここからダウンロードでき
ます 。 新しいpoissonディレクトリーで、qmakeと入力してからmakeします。 コンパイルされたポアソンプログラムは、各遺伝子の転写開始部位の背後にある500bpのウィンドウの重要度テーブルを作成します。注釈はブラウザーのゲノムから選択されます。 別の記事でブラウザーの遺伝子に関するより詳細な作業を検討します。 次に、ウィンドウに入る.bamファイルからの読み取りが選択され、それらのポアソン分布が構築されます。 結果はファイルに書き込まれます。
PoissonHandler.hpp #ifndef PoissonHandler_H #define PoissonHandler_H #include <config.hpp> class PoissonHandler : public QState { Q_OBJECT private: GenomeDescription *sql_input; GenomeDescription *sam_input; QSettings setup; public: PoissonHandler(GenomeDescription *sql,GenomeDescription *sam,HandledData &output,QState *parent=0); ~PoissonHandler(); protected: virtual void onEntry(QEvent* event); }; #endif //
PoissonHandler.cpp #include "PoissonHandler.hpp"
これらの小さなアルゴリズムは、
ZINBAや
SICERなどのより複雑なアルゴリズムの実装に
役立ちます。 それらについては別の記事で説明します。 多くの場合、タスクは注釈を使用せずに充実した領域を見つけることです。 そして、ステップバイステップで、ウィンドウに落ちてフィルターを通過したデータを選択する必要があります。
鎖特異的RNA配列決定法の包括的な比較分析Natメソッド。 2010年9月。 7(9):709–715。
ジョシュア・Z・レビン、1.6モラン・ヤスール、1,2,3,6シアン・アディコニス、1チャド・ヌスバウム、1ドーン・アン・トンプソン、1ニル・フリードマン、3,4アンドレアス・グニルケ、1、アビブ・レジェフ1,2,5
www.ncbi.nlm.nih.gov/pmc/articles/PMC3005310ChIP-Seqによるゲノム位置分析J Cell Biochem。 2009年5月1日; 107(1):11-8。
Artem Barski *およびKeji Zhao *
分子免疫学研究所、国立心臓、肺および血液研究所、NIH、ベセスダ、MD 20892
www.ncbi.nlm.nih.gov/pubmed/19173299次世代シーケンシングデータから鋭いピークと幅広いピークの両方を識別するための統合戦略。ゲノムバイオ 2011年7月25日; 12(7):120。
eng W、ha K.
ジョージワシントン大学物理学部、725 21st Street NW、ワシントンDC 20052、米国。 wpeng@gwu.edu
www.ncbi.nlm.nih.gov/pubmed/21787381ヒストン修飾ChIP-Seqデータから濃縮ドメインを識別するためのクラスタリングアプローチ。バイオインフォマティクス。 2009 8月1日; 25(15):1952-8。 Epub 2009 6月8。
Zang C、Schones DE、Zeng C、Cui K、Zhao K、Peng W.
ジョージ・ワシントン大学、ワシントンDC 20052、米国物理学部。
www.ncbi.nlm.nih.gov/pubmed/19505939ZINBAは、ローカルの共変量をDNA-seqデータと統合して、増幅されたゲノム領域内であっても、濃縮の広く狭い領域を識別します。ゲノムバイオ 2011年7月25日; 12(7):R67。
Rashid NU、Giresi PG、Ibrahim JG、Sun W、Lieb JD。
ノースカロライナ大学チャペルヒル校、チャペルヒル、ノースカロライナ州27599、米国、ギリングス国際公衆衛生学部、生物統計学科。
www.ncbi.nlm.nih.gov/pubmed/21787385レビューは、オハイオ州シンシナティのアンドレイ・カルタショフによって作成されました。